Detecting selection in low-coverage high-throughput sequencing
By A Mystery Man Writer
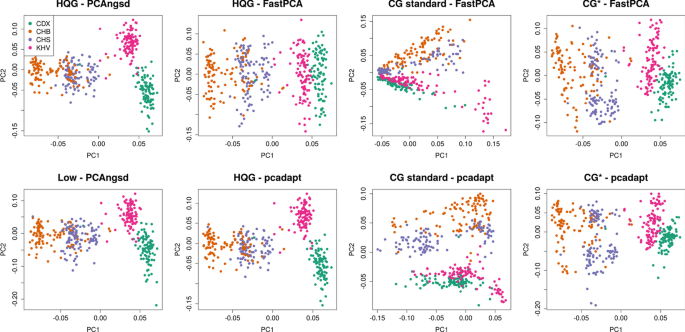
Background Identification of selection signatures between populations is often an important part of a population genetic study. Leveraging high-throughput DNA sequencing larger sample sizes of populations with similar ancestries has become increasingly common. This has led to the need of methods capable of identifying signals of selection in populations with a continuous cline of genetic differentiation. Individuals from continuous populations are inherently challenging to group into meaningful units which is why existing methods rely on principal components analysis for inference of the selection signals. These existing methods require called genotypes as input which is problematic for studies based on low-coverage sequencing data. Materials and methods We have extended two principal component analysis based selection statistics to genotype likelihood data and applied them to low-coverage sequencing data from the 1000 Genomes Project for populations with European and East Asian ancestry to detect signals of selection in samples with continuous population structure. Results Here, we present two selections statistics which we have implemented in the PCAngsd framework. These methods account for genotype uncertainty, opening for the opportunity to conduct selection scans in continuous populations from low and/or variable coverage sequencing data. To illustrate their use, we applied the methods to low-coverage sequencing data from human populations of East Asian and European ancestries and show that the implemented selection statistics can control the false positive rate and that they identify the same signatures of selection from low-coverage sequencing data as state-of-the-art software using high quality called genotypes. Conclusion We show that selection scans of low-coverage sequencing data of populations with similar ancestry perform on par with that obtained from high quality genotype data. Moreover, we demonstrate that PCAngsd outperform selection statistics obtained from called genotypes from low-coverage sequencing data without the need for ad-hoc filtering.
GitHub - Rosemeis/pcangsd: Framework for analyzing low depth NGS data in heterogeneous populations using PCA.
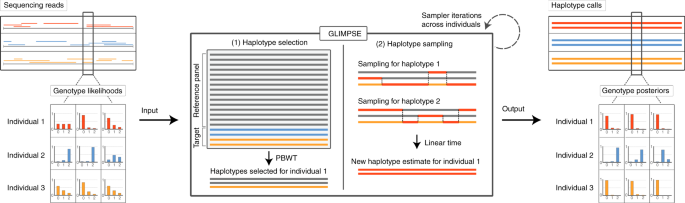
Efficient phasing and imputation of low-coverage sequencing data using large reference panels
JCM, Free Full-Text

Recent natural selection conferred protection against schizophrenia by non-antagonistic pleiotropy
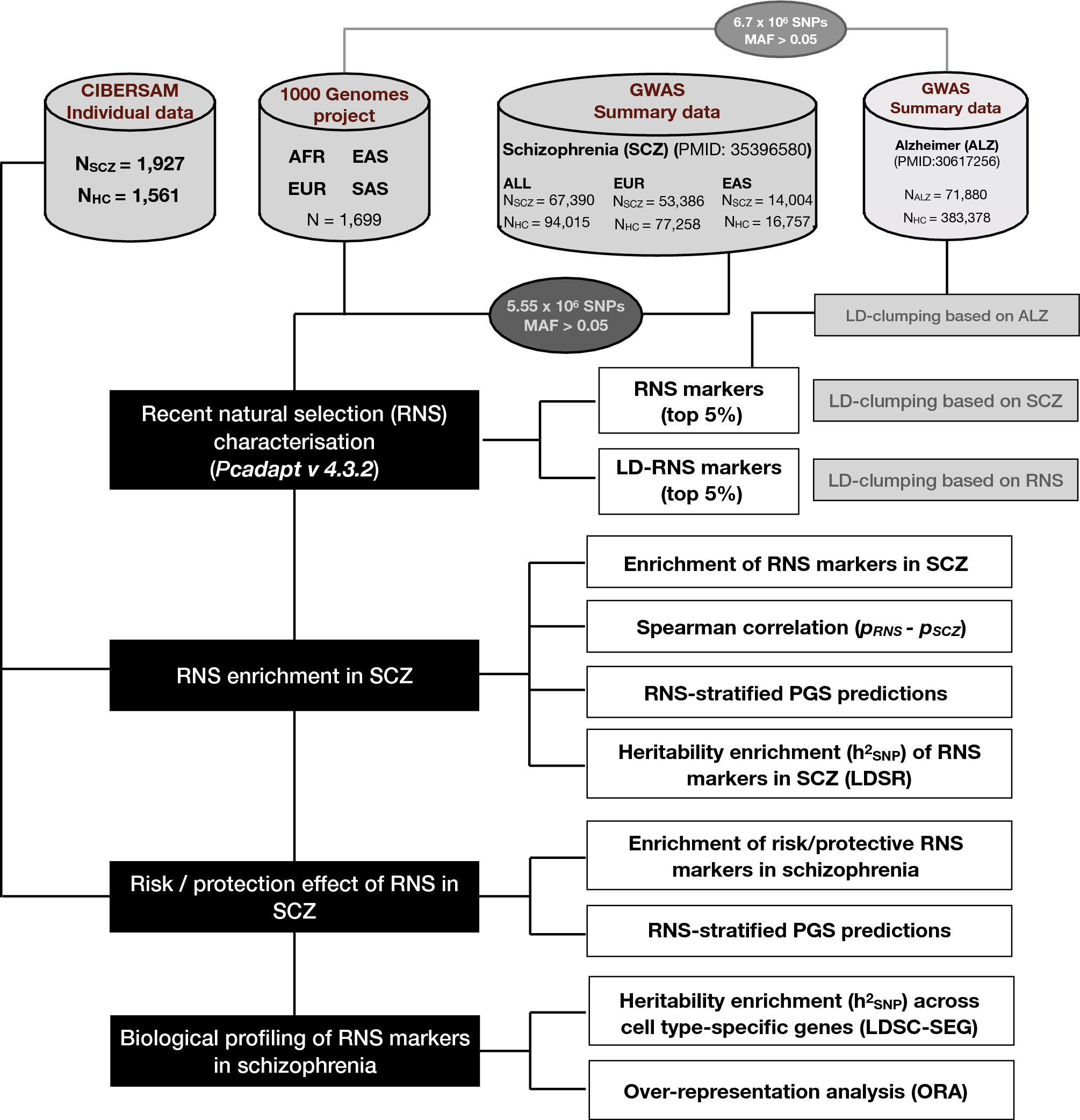
Recent natural selection conferred protection against schizophrenia by non-antagonistic pleiotropy
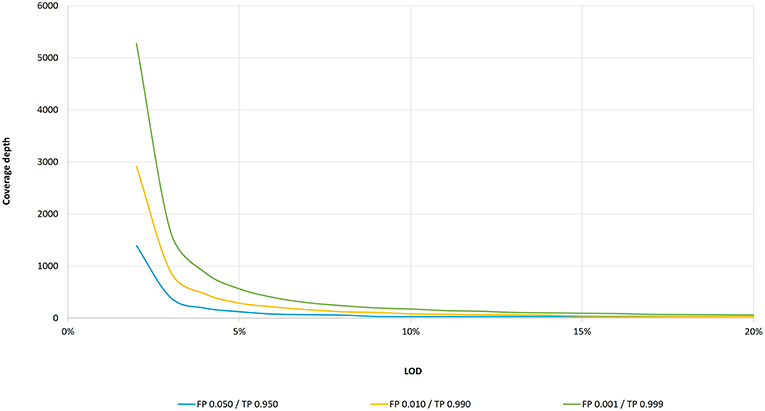
Frontiers Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics
Different approaches for high-throughput sequencing (HTS) sample

Anders ALBRECHTSEN, Professor (Associate), PhD, University of Copenhagen, Copenhagen, Bioinformatics Centre

Low-and high-throughput (NGS) IG/TR analyses in different sample types.
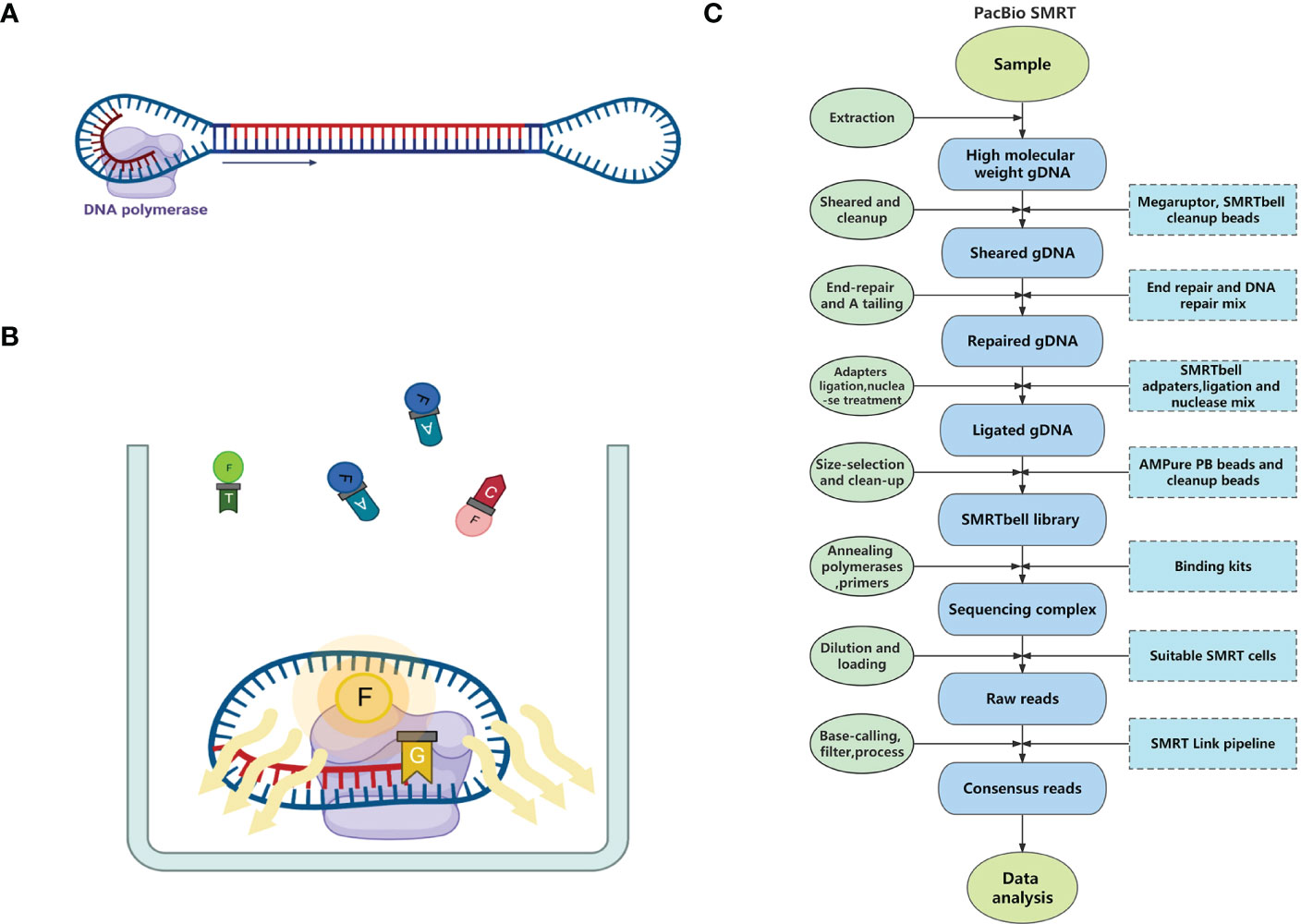
Frontiers Application of third-generation sequencing to herbal genomics

Jonas Meisner (@Rosemeis91) / X

PDF) Convergent genomic signatures of local adaptation across a continental-scale environmental gradient

Overview of High Throughput Sequencing Technologies to Elucidate Molecular Pathways in Cardiovascular Diseases
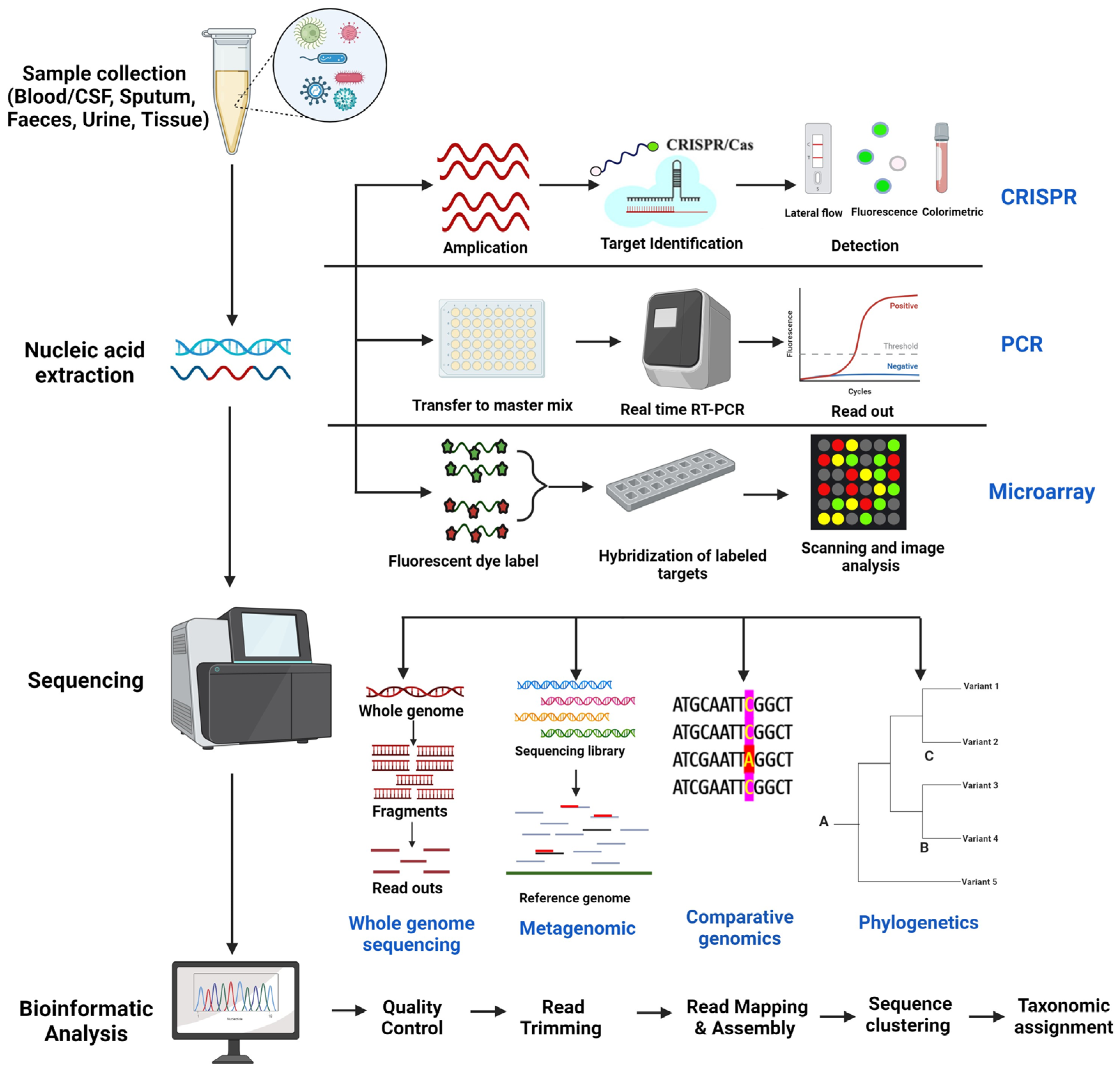
BioMedInformatics, Free Full-Text
- Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex
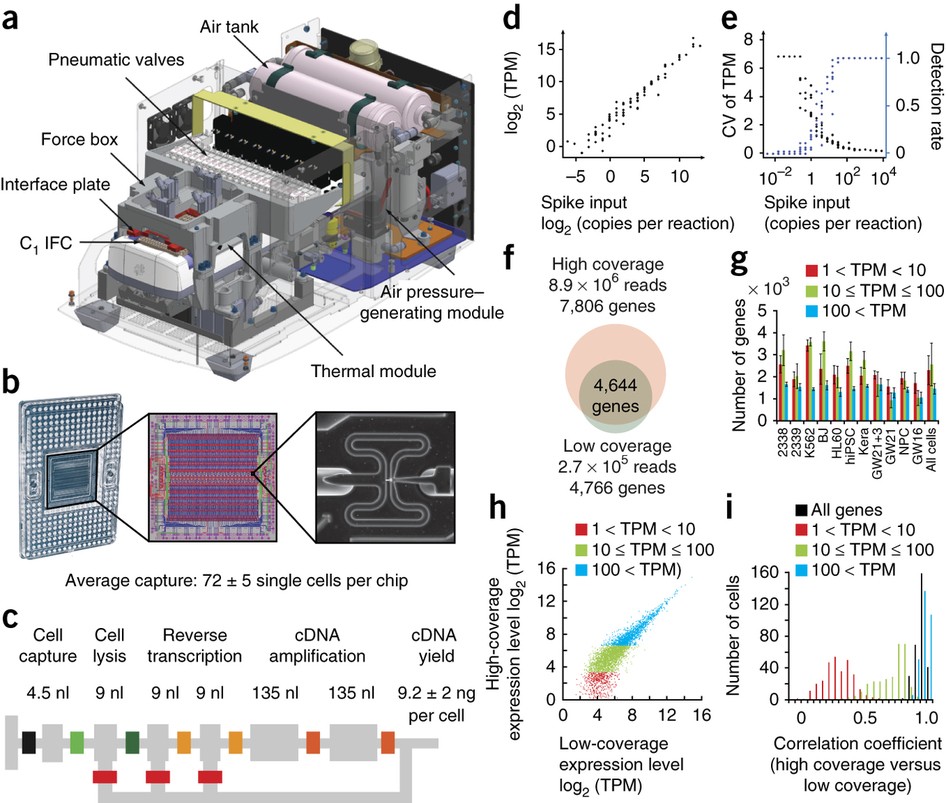
- Is connection in rural CA bad? : r/Starlink
- Experts report record low Great Lakes ice coverage

- Buy (Page 3) Zivame low coverage bra online at Zivame

- Jacoco reports low coverage with Compose tests · Issue #1208
- Buy Wholesale China Reusable Backless Self-adhesive Invisible Underwear Silicone Sticky Bra & Adhesive Bra at USD 1.59

- 32 Degrees Heat Men's Performance Jogger : : Clothing, Shoes & Accessories

- cute-workout-wear-for-less - Pretty Extraordinary

- Wrangler Retro Women's Mae Lilibeth Mid Rise Bootcut Jean 112317228

- SPRINTER Heather Grey - Jogger Pants
